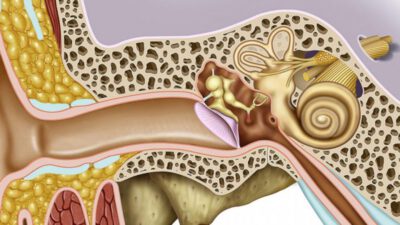
Genetik Hastalıklar
Gen değşinimlerini, kromozom yapısındaki ya da sayısındaki anormallikleri yansıtan ve işlevsel ya da anatomik değişikliklerle sonuçlanan kalıtımla aktarılmış bozukluklar. Tavşandudağı gibi biçim bozuklukları, fenilketonüri (idrarda fenilketon bulunması) gibi metabolizma bozuklukları ve albinoluk (akşınlık) yaygın görülen genetik hastalıklardır. 1935 – 1985 arasındaki yarım yüzyıl içinde enfeksiyon hastalıklarından, özellikle ishalden ölen bebeklerin oranı Batı ülkelerinde % 25′ten % 3′e inmiş, buna karşılık doğuştan oluşum bozukluklarından ötürü ölen bebeklerin oranı % 5′ten % 15′e çıkmıştır. ABD’de kromozom bozuklukları sıklığının yaklaşık 200 canlı doğumda 1 kadar olduğu belirlenmiştir. Bilinen kendiliğinden düşüklerin yaklaşık % 50-6O’ı kromozom bozukluklarına bağlıdır ve her 100 yenidoğmuş bebek ölümlerinden 6’sı, kromozom bozukluklarıyla birlikte görülmektedir.

AİLELERDE GEN AKTARIMI
Ailelerde gen aktarımı ya da kalıtım, çoğunlukla değişmiş bir genin işleviyle özdeşleşmiştir. Genetik hastalıklar, normal özelliklerin kalıtım yoluyla aktarılmasına benzer bir biçimde, kalıtımla aktarılabilir. Bu hastalıklar, otozoma bağlı baskın, eşeye bağlı baskın, otozoma bağlı çekinik ya da eşeye bağlı çekinik olan tek gen bozukluklarını içerirler. Ayrıca, çoğunlukla çevresel etkenlerle etkileşen birden çok genden kaynaklanan çok etkenli bozuklukları da kapsarlar. Otozom, bir kromozomda, eşey kromozomlarının dışındaki bir kromozom çiftinde bulunan gen çifti anlamına gelir.
TEK GEN BOZUKLUKLARI
Tek bir genin değşiniminin (mutasyonunun) neden olduğu bozukluklar, biyokimyasal yoldaki sapmaları yansıttıkları için, çoğunlukla “doğuştan metabolizma hastalıkları” diye adlandırılır. Tek bir genin değşinime uğraması sonucu olarak, normal durumlarda üretilmekte olan gen ürünü ya yoktur ya da düşük miktarlardadır. Dolayısıyla, ya önemli bir son ürünün bireşimi yeterli miktarlarda yapılamamakta ya da zehirleyici olabilen aşırı miktarlarda ara ürün birikimi oluşmaktadır. Doğuştan metabolizma hastalıklarının birçoğu, erken çocukluk döneminde öldürücü olurlar ya da gerekli beden işlevinin sürdürülmesini olanaksız kılmasalar bile,zorlaştırırlar.
Otozoma bağlı genler: Binden çoğu tam olarak belirlenmiş olan otozoma bağlı baskın genler, hem heterozigot hem de homozigot olan bireylerde görülür. Birey homozigot olursa, bunlardan birçoğunun öldürücü olduğu saptanır. Baskın özellikler genellikle hem erkekte, hem de kadında eşit olarak ortaya çıkar. Bu nedenle, anne ya da babadan biri hastalıktan etkilenmişse, her doğum için % 50 aktarılma riski bulunur. Hastalığın evriminde, gene anneden mi, babadan mı aktarılmış olduğu da rol oynayabilir .Buna Huntington koresi örnek gösterilebilir. Bu hastalık ardı arkası kesilmeyen kasılma hareketleri ve bunamayla gelişip, ölümle sonuçlanır. Belirtileri, genel olarak 35 yaşından sonra, yani çoğunlukla hastaların % 50’sinin hastalığı kalıtımla aktaracakları çocuklarının olmasından sonra ortaya çıkar.
Otozoma bağlı çekinik genler: 600′ü tam olarak belirlenmiş olan otozoma bağlı çekinik özellikler, fenotip olarak ancak hofnozigot bireylerde ortaya çıkar. Bu özelliklerin tümü olmasa bile, çoğu, bir tek gende oluşan ve biyokimyasal bir gelişme yolundaki tek bir aşamayı etkileyen bir değşinimin sonucudur. Otozoma bağlı çekinik özelliklerin çoğu, bireylerin dış görünüşlerinin ve genel sağlık durumlarının normal olmasına karşılık, heterozigot bireylerde bir ölçüye kadar ortaya çıkar. Bu kişiler, söz konusu hastalığın kendisini göstereceği çocuklarına iletebileceklerinden, “taşıyıcı” diye adlandırılırlar. Genellikle, hastalıktan, etkilenen çocukların, etkilenmemiş taşıyıcılar olan anne-babaları vardır.
Otozoma bağlı çekinik bozuklukları çocuklarına çoğunlukla, birbirleriyle çok yakın akraba olan anne-babalar aktarırlar. Heterozigot anne-babalar bakımından bu bozuklukların çocuklarında ortaya çıkma riski, her gebelik için % 25′tir.
Tay-Sachs hastalığı, galaktozemi ve fenilketonüri, otozoma bağlı çekinik hastalıklara örnek gösterilebilir. Tay-Sachs hastalığında, hasta gen, lipitlerin metabolizmada kullanılması için gerekli enzimi üretmez. Bunun sonucu olarak, bunlar beyinde oluşarak sinirsel bozukluklara ve sonunda ölüme yol açarlar. Galaktozemide, süt şekeri galaktozu glikoza çaviren enzim üretilmez ya da yeterince üretilmez. Bu bozuklukla doğan bebekler, sütle beslendiklerinde, galaktoz kanda birikir ve katarakt, karaciğer sirozu, zekâ geriliğine yol açar. Bu bozukluk, bebeğin beslenme rejiminden sütün ve süt ürünlerinin çıkarılmasıyla tedavi edilebilir. Fenolketonürili çocuklar, penilalanin aminoasitinin metabolizmasında görev alan bir enzimi üretme yeteneğinden yoksundurlar. Hastalık tedavi edilmezse, bu tür aşırı birikimler deride pigment eksikliğine ve zekâ geriliğine yol açarlar. Ortama bağlı öbür hastalıklar arasında Akdeniz kansızlığı ve orak hücre kansızlığı sayılabilir. Eşeye bağlı çekinik genler. 125′i tam olarak belirlenmiş olan eşeye bağlı çekinik özellik olgularının çoğunda anne, heterozigot olmasına karşın, etkilenmez; taşıyıcıdır. Böyle bir annenin, gen değşinimini taşıyan bir X kromozomunun geçmesiyle hasta oğullar doğurma olasılığı % 5O’dir. Bu annenin kız çocuklarının anneleri gibi heterozigot olmaları olasılığı % 5O’dir. Hasta bir erkek, üreme yeteneği varsa ve homozigot normal bir kadınla evlenirse, çocuklarından hiçbiri hasta olmaz; ama bütün kızları eşeye bağlı gen bakımından heterozigot olurlar. Kas körelmesi ve kanama hastalığı (hemofili) eşeye bağlı çekinik bozukluklara örnek gösterilebilir. Kas körelmesinde, erkek çocukların kasları, el ve ayaklardan başlayarak bütün bedene yayılıcı biçimde zayıflar ve yozlaşır. Kanama hastalığı anneleri hastalıktan etkilenmeyip yalnızca taşıyıcı olan erkek çocukları etkileyen, şiddetli kanamalarla,kanın pıhtılaşmamasıyla, yaraların iyileşmemesiyle belirti veren bir hastalıktır.
Eşeye bağlı baskın genler: Eşeye bağlı baskın genlerden kaynaklanan hastalıklardan, ayakların aşırı kıvrılarak yay biçimini almasına neden olan D vitaminine dirençli raşitizm gibi birkaçı bilinmektedir. Değşinimler X kromozomunda oluşmaktadır; o yüzden de değşinimi anne taşıyorsa, hem erkek, hem de kız çocuklarda hastalığın görülme riski % 5O’dir. Bozukluğu taşıyan babaysa, erkek çocuklarda hastalığın görülmemesine karşılık, hastalık kız çocukların tümünde ortaya çıkmaktadır.
ÇOK ETKENLİ KALITIM:
Tek değil, birkaç genin etkinliğini yansıtan bozukluklar, “çok etkenli hastalıklar” diye adlandırılır. Nispeten yaygın sayılabilecek birkaç hastalık bu sınıfa|girer:Tavşan dudağı ve damak yarıklığı, mide kapısı darlığı ve yarık omurga (Spina Bifida), vb. Genellikle, hasta anne-babaların hasta bir çocukları olması riski % 3-5 arasındadır. Anne-babadan yalnızca biri etkilenmişse de, her doğum için bu risk % 3-5 arasındadır.
KROMOZOM ANORMALLİKLERİ:
Doğuştan bozukluklarda gün geçtikçe artan oranda kromozom anormallikleri saptanmaktadır. En yaygın rastlanan anormallik, toplam kromozom sayısındaki değişmedir. Genellikle, toplam otozom sayısının azalması yaşamla bağdaşmaz. Herhangi bir otozom çiftini içeren bir fazla kromozomu bulunan bir bebekte de, bedensel anormallikler ve zekâ geriliğinin yanı sıra, yaşam süresi de sınırlı olur.
En yaygın kromozom anormalliği, mongolizmdir (Down sendromu da denir). Hastalık 21 sayısıyla gösterilen kromozomdan ileri gelir. Bu anormallik, “Trizomi 21″ diye adlandırılır; çünkü bebeğin bedenindeki bütün hücrelerde 21. çiftte fazladan bir kromozom bulunur. Hastalık 800 canlı doğumdan 1′inde görülür. Mon-golizmin ortaya çıkmasında annelik yaşı bir etken oluşturur; sözgelimi 30-40 yaş arasındaki 300 anneden biri, 40 yaşının üstünde olan 40 anneden biri, mongol bebekler doğururlar. İyi tanımlanmış öbür trizomiler, Trizomi 13 (Patau sendromu) ve Trizomi 18′dir (Edvvard sendromu). Her iki anormallik de, çocuklarda zekâ geriliğinin ve sınırlı yaşam süresinin bulunduğu çoğul doğuştan hastalıklarla birlikte görülür. Bütün insan kromozom anormalliklerini, gelişmekte olan dölütte belirlemek olasılığı vardır. Bu inceleme, gebeliğin 9. haftası kadar erken bir dönemde yapılabilen koryon villüs örneklemesiyle ya da 14.-16. hafta kadar erken bir dönemde yapılabilen amniyosentez yoluyla gerçekleştirilir.
Otozoma bağlı anormallikler gibi, eşey kromozomlarındaki normal dışılıklar da, bozulmuş üretkenlikle ya da kısırlıkla sonuçlanabilir. Sözgelimi 45 X ya da Turner sendromu adı verilen eşey kromozom anormalliği, kadınlarda görülür. Hastaların çoğu kısa boyludur; üreme organlarında ve memeler, beden kıllarının dağılımı gibi ikincil cinsellik organlarında gelişme bozuklukları görülür; ayrıca kısır olurlar. Aynı biçimden bir başka anormallik olan, Klinefelter sendromu, kişinin beden hücrelerinde XXY eşey kromozomu biçiminde, 47 kromozom bulunduğu zaman ortaya çıkar. Genel olarak erkek görünümünde, uzun boylu ve bedeni üstünde kıt kıl dağılımı olan bu kişiler, kısırdır.
Öbür eşey kromozomu anormalliklerinden hangisi bulunursa bulunsun, Y kromozomunun bulunduğu durumlarda kişi, hemen her zaman beden görünüşü bakımından erkektir. Var olan eşey kromozomlarının toplam sayısı arttıkça, doğuştan bozuklukların ve zekâ geriliğinin yaygınlığı da artar.
GEN TEDAVİSİ
Yeniden birleştirilmiş DNAteknolojisinin amaçlarından biri, genetik temeU dayanan insan hastalıklarının tedavisidir. Gen tedavisi şu gen yerleştirme tekniklerinden birini ya da daha ço ğunu kapsayabilir: (1) Gen değiştirme tedavisi{bu teda vide değşinik genin yerine normal gen yerleştirilir. Bı tür gen tedavisi, belirli bir kromozomun özel bir nokta sında bulunan genin bulunduğu yer, o genin gerekli biçimde işlev görmesi için zorunluysa önem taşır); (2) Gen artırma tedavisi (bu tedavide, normal biçimdeki bir gen, hücrenin kromozomlarından birine, anormal gen çıkarılmadan yerleştirilir. Bu tedavi, genetik hastalığa, yok olan, küçülen ya da etkisizleşen bir gen yol açmışsa etkili olur); (3) Gen etkisizleştirme tedavisi (bu tedavide, aktarılan gen, ya değşinime uğramış bir genin oluşturduğu kusurlu bir proteini ya da kendisinin birçok kopyasını verecek biçimde anormal olarak kopyalanan bir gen tarafından oluşturulan aşırı sayıda proteini yansızlaştırır). Tedavi genleri bir hücreye, hücre zarını geçici olarak yabancı DNA’ya geçirgen kılan çeşitli kimyasal ya da fiziksel işlemlerden yararlanılarak doğrudan doğruya sokulabilir. Bu aktarım yöntemine transfeksiyon adı verilir.
Dolaylı bir aktarım yöntemi olan ve transdüksiyon adı verilen yöntemse, yararlı bir geni, bir virüsün genetik gereciyle birleştirir; ardından, virüsün hedef hücreye yerleşmesine olanak verilir. Gen aktarımının en verimli yollarından birinin “retrovirüs” adı verilen bir RNA virüsünün transdüksiyonu olduğu saptanmıştır. Konak hücreye buluşmasının ardından retrovirüs, DNA’nın kendisini kopya etmesini sağlar; böylece konak hücrenin genetik gerecine yerleşir.
199O’dagenjtedavisinin|ilk sınaması,lenfositleri, bağışıklık sisteminin normal gelişmesi bakımındanıçok büyük önem taşıyan adenozin deaminaz (ADA) enzimini üretemeyen bir çocukta yapıldı. Gen artırma tedavisi uygulanarak, lennfositler çocuğun kanından çıkarıldı ve ADA geni eklenerek retrovirüs olarak değiştirildi. Ardından bu hücreler çocuğun kan dolaşımına sokuldu. Aylık kan vermelerden sonraki ilk raporlar, çocukta bağışıklık işlevinin iyileşmiş durumda olduğunu gösteriyordu.
İkinci bir gen tedavisi sınaması da, 1991 başlarında kötücül melanom deri kanserine tutulmuş iki hasta üstünde uygulanmıştır. Kanama hastalığı, kistli fibroz ve kas körelmesi gibi başka genetik hastalıkları tedavi etmekte gen tedavisinin kullanılması yolundaki çalışmalar da ilerlemektedir.
ZİYARETÇİ YORUMLARI
BİR YORUM YAZ